Nanopore ARTIC-Nanopolish SOP¶
Graphical summary¶
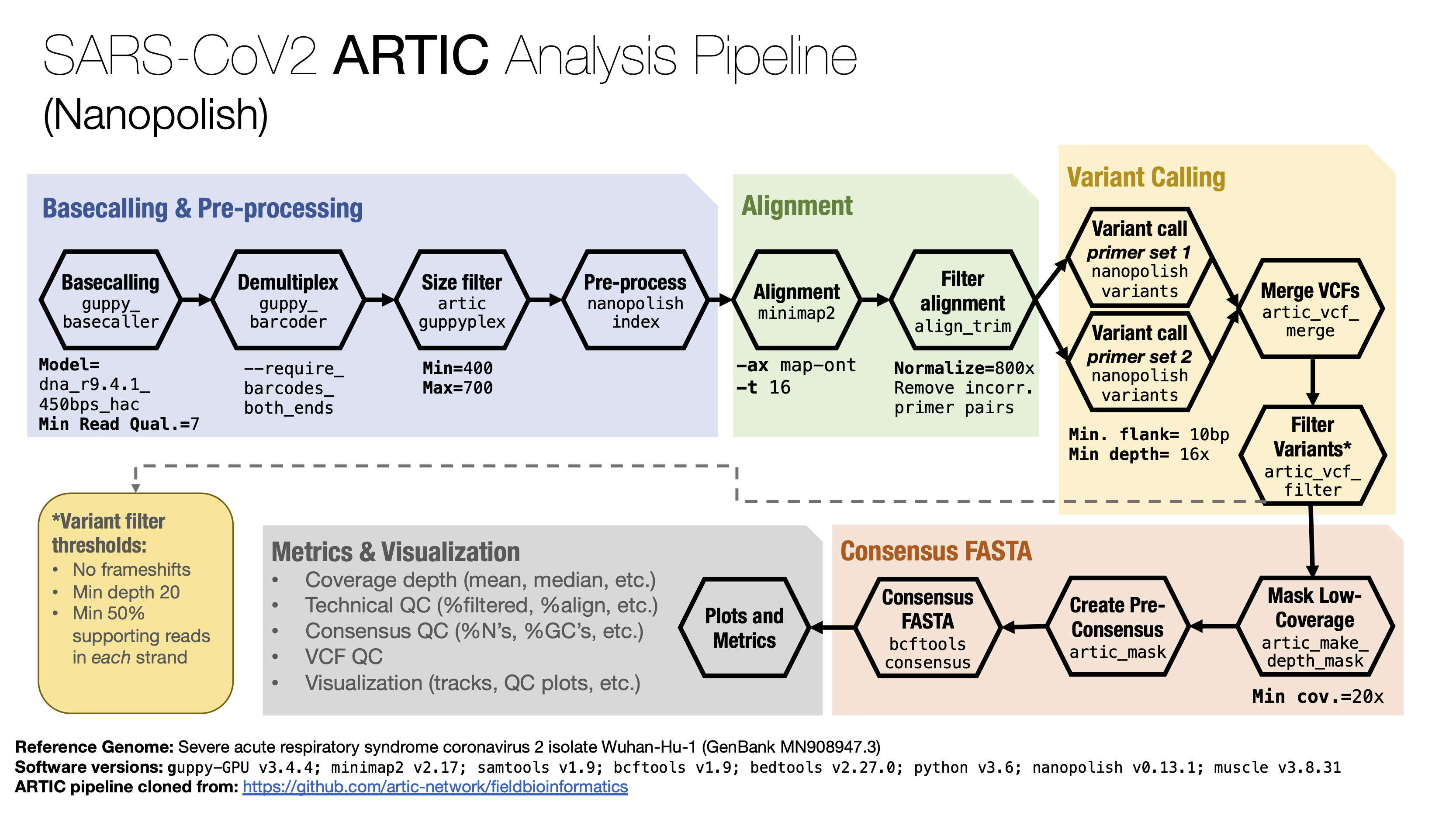
Description¶
The SOP for Nanopore data is based on the ARTIC SARS-CoV2 protocol using nanopolish. Their full documentation is found here.
The protocol was closely followed with the majority of changes, involving technical adaptations to be able to run in a High Performance Computing environment where the usage Conda is not advisable.
Steps¶
Base calling with Guppy
Raw reads are base-called with Guppy using the High Accuracy Model (flip-flop) and a minimun quality
threshold of 7:
SAMPLE="run_name"
INPUT="/path/to/fast5/"
OUTPUT="/path/to/fastq/"
PROTOCOL="dna_r9.4.1_450bps_hac.cfg"
MINQ=7
GPUPARAMS=""
RDSPERFILE=4000
OTHER_OPTIONS=""
guppy_basecaller --input_path ${INPUT} \
--recursive \
--device auto \
--save_path ${OUTPUT} \
--config ${PROTOCOL} \
--qscore_filtering --min_qscore ${MINQ} \
--compress_fastq ${GPUPARAMS} \
--records_per_fastq ${RDSPERFILE} \
--disable_pings ${OTHER_OPTIONS} \
--verbose |& tee -a ${OUTDIR}/${SAMPLE}.guppy.log
Demultiplexing with Guppy
Reads were demultiplexed using Guppy. The option --require_barcodes_both_ends is activated. Since
regular nanopore barcodes were used, the demultiplex command points to the nanopore barcode config files.:
INPUT="/path/to/fastq/"
OUTPUT="/path/to/demultiplex/"
guppy_barcoder --require_barcodes_both_ends \
--input_path ${INPUT} \
--save_path ${OUTPUT} \
--arrangements_files "barcode_arrs_nb12.cfg barcode_arrs_nb24.cfg" \
ARTIC-Nanopolish pipeline
Since some HPC environments like Compute Canada don’t support python package managers like Conda, the python
environment to use a tool like the ARTIC protocol needs to be defined initially, before running the pipeline.
Compute Canada maintains several python package wheels internally, so several dependencies are already met,
however, others had to be manually downloaded and compiled. In this case, they were all deposited in a folder
saved as an environment variable called $PYTHON_WHL. Each SLURM job, then uses the following code to setup
the python environment and actiavte it:
export ENVDIR="${SLURM_TMPDIR}/env"
export PYTHON_WHL="/path/to/python/wheels/"
export ARTIC_WHL="/path/to/artic.whl"
virtualenv --no-download ${ENVDIR}
source ${ENVDIR}/bin/activate
pip install --no-index --upgrade pip
pip install --no-index biopython
pip install --no-deps ${PYTHON_WHL}/clint-0.5.1/dist/clint-0.5.1-py3-none-any.whl
pip install --no-index pandas
pip install --no-index pysam
pip install --no-index pytest
pip install --no-deps ${PYTHON_WHL}/PyVCF-0.6.8/dist/PyVCF-0.6.8-py3-none-any.whl
pip install --no-index requests
pip install --no-index tqdm
pip install --no-deps ${PYTHON_WHL}/whatshap-0.18/dist/whatshap-0.18-cp36-cp36m-linux_x86_64.whl
pip install --no-deps ${ARTIC_WHL}
3.1 Read size filtering
Using the ARTIC guppyplex tool, we filter the reads that don’t fit into the expected lengths.:
MIN_LENGTH="400"
MAX_LENGTH="700"
FASTQ_DIR="/path/to/demultiplex/fatsq"
POOL="pool_name"
artic guppyplex \
--min-length ${MIN_LENGTH} \
--max-length ${MAX_LENGTH} \
--directory ${FASTQ_DIR} \
--prefix ${POOL}
3.2. Run ARTIC Nanopolish pipeline
Run the ARTIC nanopolish pipeline using the following command:
NORMALISE="800"
THREADS="16"
PRIMERS_DIR="/path/to/pimers/dir/"
POOL="pool_name"
BARCODE="barcodeXX"
FAST5_DIR="/path/to/fast5/"
SUMMARY="/path/to/sequencing_summary.txt"
PRIMERS_VER="nCoV-2019/V3"
SAMPLE="sample_name"
artic minion \
--normalise ${NORMALISE} \
--threads ${THREADS} \
--scheme-directory ${PRIMERS_DIR} \
--read-file ${POOL}_${BARCODE}.fastq \
--fast5-directory ${FAST5_DIR} \
--sequencing-summary ${SUMMARY} \
${PRIMERS_VER} \
${SAMPLE} |& tee -a ${SAMPLE}_nanopolish.log
The above command actually runs the following tools:
nanopolish index -s ${SUMMARY} -d ${OUTDIR}
minimap2 -a -x map-ont -t 16 primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta ${POOL}_${BARCODE}.fastq | samtools view -bS -F 4 - | samtools sort -o ${SAMPLE}
samtools index ${SAMPLE}.sorted.bam
align_trim --start --normalise 800 primer_schemes/nCoV-2019/V3/nCoV-2019.scheme.bed --report ${SAMPLE}.alignreport.txt < ${SAMPLE}.sorted.bam 2> ${SAMPLE}
align_trim --normalise 800 primer_schemes/nCoV-2019/V3/nCoV-2019.scheme.bed --remove-incorrect-pairs --report ${SAMPLE}.alignreport.txt < ${SAMPLE}.sorted
samtools index ${SAMPLE}.trimmed.rg.sorted.bam
samtools index ${SAMPLE}.primertrimmed.rg.sorted.bam
nanopolish variants --min-flanking-sequence 10 -x 1000000 --progress -t 16 --reads ${POOL}_${BARCODE}.fastq -o ${SAMPLE}.nCoV-2019_2.vcf -b ${SAMPLE}.trimmed.rg.sorted.bam -g /lustre03/project/6007512/C3G/projects/Moreira_COVID19_Genotyping/artic
nanopolish variants --min-flanking-sequence 10 -x 1000000 --progress -t 16 --reads ${POOL}_${BARCODE}.fastq -o ${SAMPLE}.nCoV-2019_1.vcf -b ${SAMPLE}.trimmed.rg.sorted.bam -g /lustre03/project/6007512/C3G/projects/Moreira_COVID19_Genotyping/artic
artic_vcf_merge ${SAMPLE} primer_schemes/nCoV-2019/V3/nCoV-2019.scheme.bed nCoV-2019_2:${SAMPLE}.nCoV-2019_2.vcf nCoV-2019_1:${SAMPLE}.nCoV-2019_1.v
artic_vcf_filter --nanopolish ${SAMPLE}.merged.vcf ${SAMPLE}.pass.vcf ${SAMPLE}.fail.vcf
artic_make_depth_mask primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta ${SAMPLE}.primertrimmed.rg.sorted.bam ${SAMPLE}.coverage_mask.txt
bgzip -f ${SAMPLE}.pass.vcf
tabix -p vcf ${SAMPLE}.pass.vcf.gz
artic_mask primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta ${SAMPLE}.coverage_mask.txt ${SAMPLE}.fail.vcf ${SAMPLE}.preconsensus.fasta
bcftools consensus -f ${SAMPLE}.preconsensus.fasta ${SAMPLE}.pass.vcf.gz -m ${SAMPLE}.coverage_mask.txt -o ${SAMPLE}.consensus.fasta
artic_fasta_header ${SAMPLE}.consensus.fasta "${SAMPLE}/ARTIC/nanopolish"
cat ${SAMPLE}.consensus.fasta primer_schemes/nCoV-2019/V3/nCoV-2019.reference.fasta > ${SAMPLE}.muscle.in.fasta
muscle -in ${SAMPLE}.muscle.in.fasta -out ${SAMPLE}.muscle.out.fasta
Create metrics
For metrics generation, the following tools are run:
samtoolsfor general alignment stats.bedtoolsfor coverage metricsWUBfor additional alignment metrics see this repository.
REFERENCE="/path/to/SARS-CoV2/reference.fasta"
SAMPLE="sample_name"
SORTED_BAM="/path/to/${SAMPLE}.sorted.bam"
samtools view -@ 4 -F 0x900 \
--output-fmt BAM \
--reference ${REFERENCE} \
-o ${SAMPLE}.nosup.bam \
${SORTED_BAM}
samtools sort -@ 4 --reference ${REFERENCE} \
--output-fmt BAM \
-o ${SAMPLE}.nosup.sorted.bam \
${SAMPLE}.nosup.bam
samtools index -@ 5 -b ${SAMPLE}.nosup.sorted.bam
bedtools genomecov -bga -ibam ${SAMPLE}.nosup.bam > ${SAMPLE}.nosup.bedGraph
bedtools genomecov -ibam ${SAMPLE}.nosup.bam > ${SAMPLE}.nosup.histogram
samtools stats ${SAMPLE}.nosup.bam > ${SAMPLE}.nosup.bam.stats
samtools depth ${SAMPLE}.nosup.bam > ${SAMPLE}.nosup.bam.depth
python ${WUB}/scripts/bam_alignment_qc.py -f ${REFERENCE} ${SAMPLE}.nosup.sorted.bam
Plotting and reporting is done using a combination of R scripts that parse the BAM, VCF and metrics files.
Reference Genome and Software Versions¶
Reference Genome: Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1 (GenBank MN908947.3)
Software versions
guppy-GPU v3.4.4
minimap2 v2.17
samtools v1.9
bcftools v1.9
bedtools v2.27.0
python v3.6
nanopolish v0.13.1
muscle v3.8.31