Illumina ARTIC SOP¶
Graphical summary¶
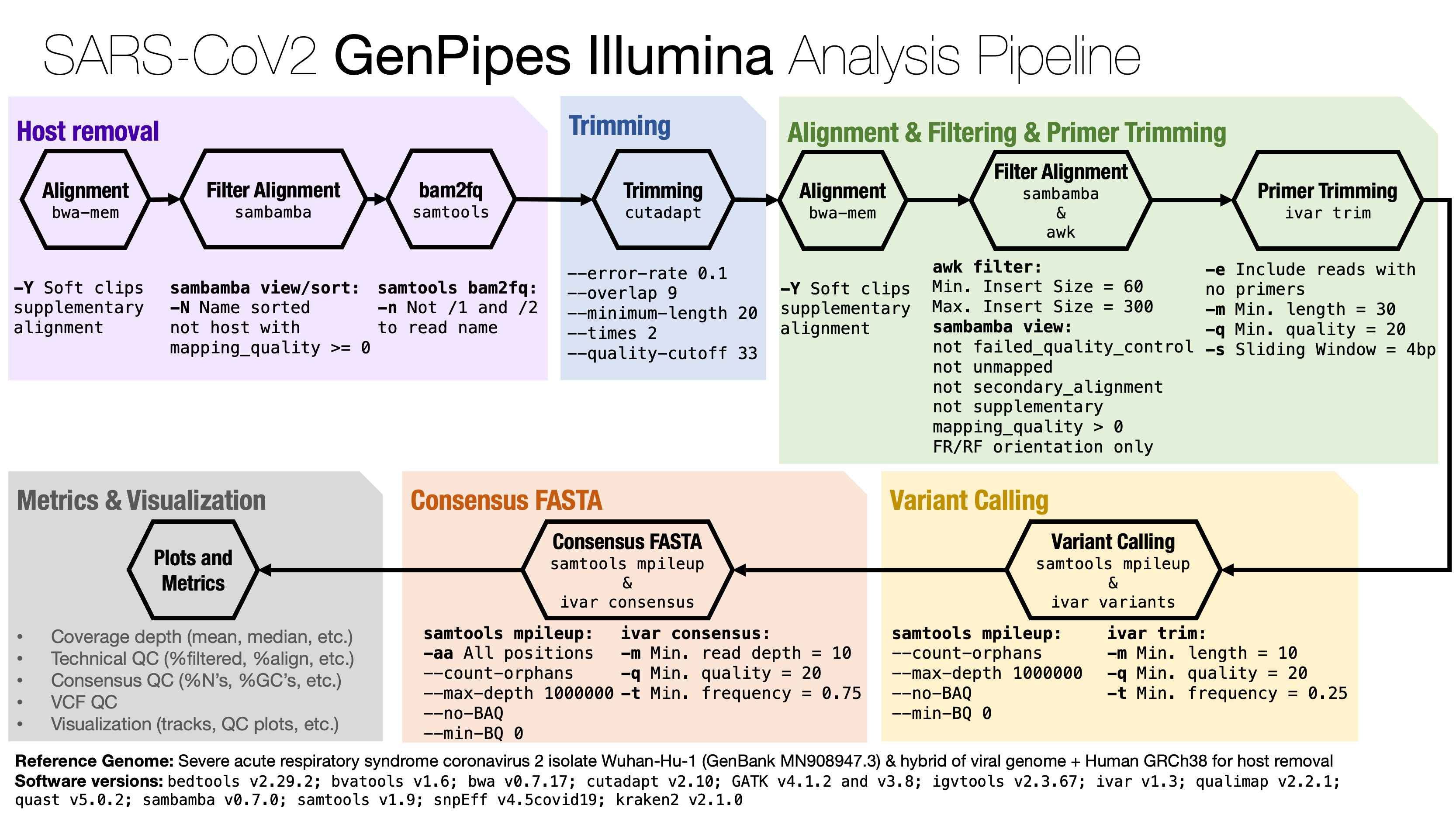
Description¶
The Illumina analysis pipeline is based on the steps suggested by Paragon Genomics, the manufacturer of the
CleanPlex amplicon panel, with important modifications to improve performance.
Steps¶
Align read with
bwa mem, filter out human reads on the bam file and convert to fastq
1.1 Raw reads are aligned with bwa mem and sorted by position with sambamba for merics
bwa mem \
-K 100000000 -v 3 -t 8 -Y \
-R '@RG\tID:${SAMPLE}\tSM:${SAMPLE}\tLB:${LIBRARY}\tPU:run1_2\tCN:McGill University and Genome Quebec Innovation Centre\tPL:Illumina' \
Coronavirinae.SARS-CoV-2/genome/bwa_index/Hybrid.SARS-CoV-2.GRCh38.fa \
data/${SAMPLE}/${SAMPLE}.pair1.fastq.gz \
data/${SAMPLE}/${SAMPLE}.pair2.fastq.gz | \
sambamba view -S -f bam \
/dev/stdin | \
sambamba sort \
/dev/stdin \
--out host_removal/${SAMPLE}/${SAMPLE}.hybrid.sorted.bam
1.2 Human mapped reads with a mapping quality > 0 are filtered out and then sorted by name with sambamba
sambamba view \
-f bam -F "not (ref_name =~ /chr*/ and mapping_quality >= 0)" \
host_removal/${SAMPLE}/${SAMPLE}.hybrid.sorted.bam | \
sambamba sort \
-N \
/dev/stdin \
--out host_removal/${SAMPLE}/${SAMPLE}.host_removed.nsorted.bam
1.3 Conversion of the bam filtered out and sorted by name into fastqs with samtools
samtools bam2fq \
-@ 10 \
-n \
-1 host_removal/${SAMPLE}/${SAMPLE}.host_removed.pair1.fastq.gz \
-2 host_removal/${SAMPLE}/${SAMPLE}.host_removed.pair2.fastq.gz \
-0 host_removal/${SAMPLE}/${SAMPLE}.host_removed.other.fastq.gz \
-s host_removal/${SAMPLE}/${SAMPLE}.host_removed.single.fastq.gz \
host_removal/${SAMPLE}/${SAMPLE}.host_removed.nsorted.bam
Trim sequencing adaptor with
cutadapt
cutadapt -g GAACGACATGGCTACGATCCGACTT \
-G GACCGCTTGGCCTCCGACTT \
-a AAGTCGGAGGCCAAGCGGTC \
-A AAGTCGGATCGTAGCCATGTCGTTC \
-j 5 \
-e 0.1 -O 9 -m 20 -n 2 --quality-cutoff 33 \
-o cleaned_raw_reads/${SAMPLE}/${SAMPLE}.trim.pair1.fastq.gz \
-p cleaned_raw_reads/${SAMPLE}/${SAMPLE}.trim.pair2.fastq.gz \
host_removal/${SAMPLE}/${SAMPLE}.host_removed.pair1.fastq.gz \
host_removal/${SAMPLE}/${SAMPLE}.host_removed.pair2.fastq.gz
Align read with
bwa memand filter the bam file
3.1 Multimapping of the reads with bwa mem and sort with sambamba
bwa mem \
-K 100000000 -v 3 -t 8 -Y \
-R '@RG\tID:${SAMPLE}\tSM:${SAMPLE}\tLB:${LIBRARY}\tPU:run1_2\tCN:McGill University and Genome Quebec Innovation Centre\tPL:Illumina' \
Coronavirinae.SARS-CoV-2/genome/bwa_index/Coronavirinae.SARS-CoV-2.fa \
trim/${SAMPLE}/${SAMPLE}.trim.pair1.fastq.gz \
trim/${SAMPLE}/${SAMPLE}.trim.pair2.fastq.gz | \
sambamba view -S -f bam \
/dev/stdin | \
sambamba sort \
/dev/stdin \
--out alignment/${SAMPLE}/${SAMPLE}.sorted.bam && \
sambamba index \
alignment/${SAMPLE}/${SAMPLE}.sorted.bam \
alignment/${SAMPLE}/${SAMPLE}.sorted.bam.bai
3.2 Mapped reads are filtered with sambamba and awk
sambamba view -h \
alignment/${SAMPLE}/${SAMPLE}.sorted.bam | \
awk 'substr($0,1,1)=="@" || ($9 >= 60 && $9 <= 300) || ($9 <= -60 && $9 >= -300)' | \
sambamba view -S -f bam -F "not failed_quality_control and not unmapped and not secondary_alignment and mapping_quality > 0 and not supplementary and not (((mate_is_reverse_strand and reverse_strand) or (not reverse_strand and not mate_is_reverse_strand)))" \
/dev/stdin \
-o alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.bam
Trim Amplicon primers with
ivarandsamtools
ivar trim -i alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.bam \
-p alignment/${SAMPLE}/${SAMPLE}.primerTrim \
-b Coronavirinae.SARS-CoV-2/Coronavirinae.SARS-CoV-2.Illumina_ARTIC_primers.bed \
-f Coronavirinae.SARS-CoV-2/Coronavirinae.SARS-CoV-2.Illumina_ARTIC_primerpair.tsv \
-m 30 -q 20 -s 4 -e && \
sambamba sort \
alignment/${SAMPLE}/${SAMPLE}.primerTrim.bam \
--out alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.bam
Call variants with
ivarandsamtools
samtools mpileup -A -d 1000000 -B -Q 0 \
-l $MUGQIC_INSTALL_HOME_DEV/genomes/species/Coronavirinae.SARS-CoV-2/artic_amplicon.bed \
--reference Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
-f Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.bam | \
ivar variants -p alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim \
-r Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
-m 10 -q 20 -t 0.25 && \
ivar_variants_to_vcf.py alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.tsv alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.vcf && \
bgzip -cf \
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.vcf > \
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.vcf.gz && \
tabix -pvcf alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.vcf.gz
Annotate the variants with
SnpEff
java -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx20G \
-jar $SNPEFF_HOME/snpEff.jar ann \
-v -c $SNPEFF_HOME/snpEff.config \
-s metrics/dna/${SAMPLE}/snpeff_metrics/${SAMPLE}.snpEff.html \
MN908947.3 \
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.vcf.gz > \
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.annotate.vcf && \
bgzip -cf \
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.annotate.vcf > \
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.annotate.vcf.gz && \
tabix -pvcf alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.annotate.vcf.gz
Generate consensus FASTA and rename header with
ivarandsamtools
7.1 Consensus generation with** ivar and samtools
samtools mpileup -aa -A -d 600000 -Q 0 \
-f Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.bam | \
ivar consensus -p alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.consensus -q 20 -t 0.75 -m 10
7.2 Renaming of header for GISAID submission and consensus file for flagging outlayers (too many Ns, frameshifts and/or low coverage)
cons_len=`grep -oP "Total length \(>= 0 bp\)\t\K.*?(?=$)" metrics/dna/${SAMPLE}/quast_metrics/report.tsv`
N_count=`grep -oP "# N's\",\"quality\":\"Less is better\",\"values\":\[\K.*?(?=])" metrics/dna/${SAMPLE}/quast_metrics/report.html`
cons_perc_N=`echo "scale=2; 100*$N_count/$cons_len" | bc -l`
frameshift=`if grep -q "frameshift_variant" variant/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.annotate.vcf; then echo "FLAG"; fi`
genome_size=`awk '{print $2}' Coronavirinae.SARS-CoV-2/Coronavirinae.SARS-CoV-2.fa.fai`
bam_cov50X=`awk '{if ($4 > 50) {count = count + $3-$2}} END {if (count) {print count} else {print 0}}' alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.BedGraph`
bam_cov50X=`echo "scale=2; 100*$bam_cov50X/$genome_size" | bc -l`
STATUS=`awk -v bam_cov50X=$bam_cov50X -v frameshift=$frameshift -v cons_perc_N=$cons_perc_N 'BEGIN { if (cons_perc_N < 1 && frameshift != "FLAG" && bam_cov50X >= 90) {print "pass"} else if ((cons_perc_N >= 1 && cons_perc_N <= 5) || frameshift == "FLAG" || bam_cov50X < 90) {print "flag"} else if (cons_perc_N > 5) {print "rej"} }'`
export STATUS && \
awk '/^>/{print ">Canada/Qc-${SAMPLE}/2020 seq_method:Illumina_NexteraFlex|assemb_method:ivar|snv_call_method:ivar"; next}{print}' < consensus/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.consensus.fa > consensus/${SAMPLE}/${SAMPLE}.consensus.illumina.${STATUS}.fasta
Compute metrics
A series of scripts compute several metrics derived from the output of the analysis above. Those metrics are calculated at different steps in the pipeline and some are required for renaming the consensus file. Here are the full set of commands used to generate these metrics.:
#kraken2 metrics right after dehosting
kraken2 \
--quick \
--threads 5 \
--db kraken2-2.1.0/db \
--paired \
--unclassified-out metrics/dna/${SAMPLE}/kraken_metrics/${SAMPLE}.unclassified_sequences#.fastq \
--classified-out metrics/dna/${SAMPLE}/kraken_metrics/${SAMPLE}.classified_sequences#.fastq \
--output metrics/dna/${SAMPLE}/kraken_metrics/${SAMPLE}.kraken2_output \
--report metrics/dna/${SAMPLE}/kraken_metrics/${SAMPLE}.kraken2_report \
host_removal/${SAMPLE}/${SAMPLE}.host_removed.pair1.fastq.gz \
host_removal/${SAMPLE}/${SAMPLE}.host_removed.pair2.fastq.gz
#alignment and insert size metrics
gatk --java-options "-XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx20G" \
CollectMultipleMetrics \
--PROGRAM=CollectAlignmentSummaryMetrics \
--PROGRAM=CollectInsertSizeMetrics \
--VALIDATION_STRINGENCY=SILENT \
--REFERENCE_SEQUENCE=Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
--INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam \
--OUTPUT=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.all.metrics \
--MAX_RECORDS_IN_RAM=1000000
#OxoG metrics
gatk --java-options "-XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx20G" \
CollectOxoGMetrics \
--VALIDATION_STRINGENCY=SILENT \
--INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam \
--OUTPUT=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.oxog_metrics.txt \
--REFERENCE_SEQUENCE=Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
--MAX_RECORDS_IN_RAM=4000000
#GC biais metrics
gatk --java-options "-XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx20G" \
CollectGcBiasMetrics \
--VALIDATION_STRINGENCY=SILENT \
--ALSO_IGNORE_DUPLICATES=TRUE \
--INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam \
--OUTPUT=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.qcbias_metrics.txt \
--CHART=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.qcbias_metrics.pdf \
--SUMMARY_OUTPUT=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.qcbias_summary_metrics.txt \
--REFERENCE_SEQUENCE=Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
--MAX_RECORDS_IN_RAM=4000000
#qualimap metrics
qualimap bamqc -nt 10 \
-bam alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam -outdir metrics/dna/${SAMPLE}/qualimap \
--java-mem-size=55G
#flagstats metrics primerTrimed
sambamba flagstat \
alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam \
> metrics/dna/${SAMPLE}/flagstat/${SAMPLE}.sorted.primerTrim.flagstat
#bedGraph generation primerTrimmed
bedtools genomecov -bga \
-ibam alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam > alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.BedGraph
#hs metrics
gatk --java-options "-XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx20G" \
CollectHsMetrics \
--INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam \
--OUTPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.onTarget.tsv \
--BAIT_INTERVALS=Coronavirinae.SARS-CoV-2/SARSCoV2.ampInsert.interval_list \
--TARGET_INTERVALS=SARSCoV2.ampInsert.interval_list \
--REFERENCE_SEQUENCE=Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa
#depth of coverage
java -XX:ParallelGCThreads=2 -Xmx8000M -jar $GATK_JAR \
--analysis_type DepthOfCoverage --omitDepthOutputAtEachBase --logging_level ERROR \
--reference_sequence Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
--input_file alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam \
--out alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.all.coverage \
--intervals Coronavirinae.SARS-CoV-2/artic_amplicon.bed \
--summaryCoverageThreshold 10 \
--summaryCoverageThreshold 25 \
--summaryCoverageThreshold 50 \
--summaryCoverageThreshold 75 \
--summaryCoverageThreshold 100 \
--summaryCoverageThreshold 500 \
--start 1 --stop 500 \
--nBins 499 \
--downsampling_type NONE
java -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx31G -jar $BVATOOLS_JAR \
depthofcoverage --gc --maxDepth 1001 --summaryCoverageThresholds 10,25,50,75,100,500,1000 --minMappingQuality 15 --minBaseQuality 15 --ommitN \
--threads 8 \
--ref Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
--intervals Coronavirinae.SARS-CoV-2/artic_amplicon.bed \
--bam alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam \
> alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.coverage.tsv
#generate tdf file
java -Xmx8G -Djava.awt.headless=true -jar $IGVTOOLS_JAR count -f min,max,mean -w 25 \
alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.bam \
alignment/${SAMPLE}/${SAMPLE}.sorted.primerTrim.tdf \
Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa.fai
#generate quast metrics
quast.py -r Coronavirinae.SARS-CoV-2/genome/Coronavirinae.SARS-CoV-2.fa \
--output-dir metrics/dna/${SAMPLE}/quast_metrics \
--threads 5 \
alignment/${SAMPLE}/${SAMPLE}.sorted.filtered.primerTrim.consensus.gisaid_renamed.fa
Reference Genome and Software Versions¶
Reference Genome: Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1 (GenBank MN908947.3)
Software versions
bedtools v2.29.2
bvatools v1.6
bwa v0.7.17
cutadapt v2.10
GATK v4.1.2 and v3.8
igvtools v2.3.67
ivar v1.3
kraken2 v2.1.0
qualimap v2.2.1
quast v5.0.2
sambamba v0.7.0
samtools v1.11
snpEff v4.5covid19