Illumina/MGI DNAseq WGS SOP¶
Graphical summary¶
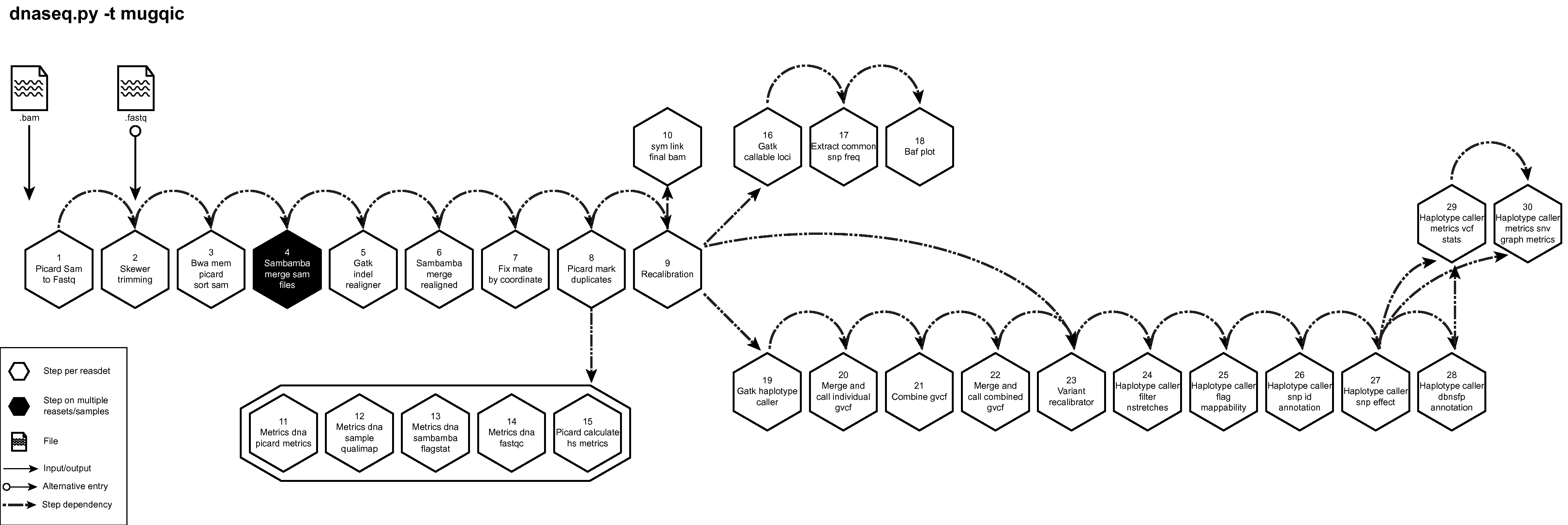
Description¶
The Illumina DNASeq WGS analysis pipeline is based on GATK best practices with important modifications to improve performance.
Steps¶
Trim sequencing adaptors with quality and size selection
skewer
1.1 Trim reads to remove low quality tail and apply size selection to remove small fragments
skewer --threads 7 --min 25 -q 25 --compress -f sanger \
-x trim/dnaseq_GM12878_chr19/adapter.tsv \
data/${SAMPLE}/${SAMPLE}.pair1.fastq.gz \
data/${SAMPLE}/${SAMPLE}.pair2.fastq.gz \
-o trim/${SAMPLE}/${SAMPLE}
Align trimmed reads with
bwa mem, and apply refinement steps to fix the heuristic of the aligner
2.1 Trimmed reads are aligned with bwa mem and sorted by position with sambamba for downstream refinement steps
bwa mem \
-M -t 15 \
-R '@RG\tID:${SAMPLE}tSM:${SAMPLE}\tLB:${LIBRARY}\tPU:run1_1\tCN:McGill University and Genome Quebec Innovation Centre\tPL:Illumina' \
Homo_sapiens.GRCh38/genome/bwa_index/Homo_sapiens.GRCh38.fa \
data/${SAMPLE}/${SAMPLE}.pair1.trim.fastq.gz \
data/${SAMPLE}/${SAMPLE}.pair2.trim.fastq.gz | \
sambamba view -S -f bam \
/dev/stdin | \
sambamba sort \
/dev/stdin \
--out alignment/${SAMPLE}/${SAMPLE}.sorted.bam && \
sambamba index \
alignment/${SAMPLE}/${SAMPLE}.sorted.bam \
alignment/${SAMPLE}/${SAMPLE}.sorted.bam.bai
2.2 Corrections to variant flanking indels is improved by doing local reassembly of regions surrounding indels
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:+UseParallelGC -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx6G -jar $GATK_JAR \
--analysis_type RealignerTargetCreator -nct 1 -nt 3 \
--reference_sequence Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--input_file alignment/${SAMPLE}/${SAMPLE}.sorted.bam \
--known Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.Mills_and_1000G_gold_standard.indels.vcf.gz \
--out alignment/${SAMPLE}/realign/all.intervals && \
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:+UseParallelGC -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx12G -jar $GATK_JAR \
--analysis_type IndelRealigner -nt 1 -nct 1 \
--reference_sequence Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--input_file alignment/${SAMPLE}/${SAMPLE}.sorted.bam \
--targetIntervals alignment/${SAMPLE}/realign/all.intervals \
--knownAlleles Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.Mills_and_1000G_gold_standard.indels.vcf.gz \
--out alignment/${SAMPLE}/realign/all.bam \
--maxReadsInMemory 750000 && \
ln -s -f \
alignment/${SAMPLE}/realign/all.bam \
alignment/${SAMPLE}/${SAMPLE}.sorted.realigned.bam
2.3 Marking of duplicate with picard
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:+UseParallelGC -XX:ParallelGCThreads=2 -Dsamjdk.buffer_size=4194304 -Xmx16G -jar $PICARD_HOME/picard.jar \
MarkDuplicates \
REMOVE_DUPLICATES=false VALIDATION_STRINGENCY=SILENT CREATE_INDEX=true \
INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.realigned.bam \
OUTPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
METRICS_FILE=alignment/${SAMPLE}/${SAMPLE}.sorted.dup.metrics \
MAX_RECORDS_IN_RAM=1000000
2.4 Base recalibration (BSQR) to detects and adjust base qualities of systematic errors made by the sequencing machine when it estimates the accuracy of each base call
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=1048576 -Xmx55G -jar $GATK_JAR \
--analysis_type BaseRecalibrator --bqsrBAQGapOpenPenalty 30 \
-nt 1 --num_cpu_threads_per_data_thread 12 \
--input_file alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
--reference_sequence Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--knownSites Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.dbSNP142.vcf.gz \
--knownSites Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.gnomad.exomes.r2.0.2.sites.no-VEP.nohist.tidy.vcf.gz \
--knownSites Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.Mills_and_1000G_gold_standard.indels.vcf.gz \
--out alignment/${SAMPLE}/${SAMPLE}.sorted.dup.recalibration_report.grp
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:+UseParallelGC -XX:ParallelGCThreads=4 -Dsamjdk.buffer_size=4194304 -Xmx30G -jar $GATK_JAR \
--analysis_type PrintReads --generate_md5 \
-nt 1 --num_cpu_threads_per_data_thread 5 \
--input_file alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--BQSR alignment/${SAMPLE}/${SAMPLE}.sorted.dup.recalibration_report.grp \
--out alignment/${SAMPLE}/${SAMPLE}.sorted.dup.recal.bam
Germline variant calling using
GATK Haplotype caller
3.1 Variant calling is performed on individual samples in padded vcf mode (gVCF). Typically run by chromosome or groups of chromosomes depending on coverage of sequenced sample whereby higher coverage approaches one chromosome per job.
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=1048576 -Xmx30G -jar $GATK_JAR \
--analysis_type HaplotypeCaller --useNewAFCalculator --emitRefConfidence GVCF -dt none -nct 1 -G StandardAnnotation -G StandardHCAnnotation \
--disable_auto_index_creation_and_locking_when_reading_rods \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--input_file alignment/${SAMPLE}/${SAMPLE}.sorted.dup.recal.bam \
--out alignment/${SAMPLE}/rawHaplotypeCaller/${SAMPLE}.hc.g.vcf.gz
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=1048576 -Xmx30G -jar $GATK_JAR \
--analysis_type GenotypeGVCFs --useNewAFCalculator -G StandardAnnotation -G StandardHCAnnotation \
--disable_auto_index_creation_and_locking_when_reading_rods \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--variant alignment/${SAMPLE}/rawHaplotypeCaller/${SAMPLE}.hc.g.vcf.gz \
--out alignment/${SAMPLE}/${SAMPLE}.hc.vcf.gz
3.2 Joint calling of multiple N samples is preformed by GATK combineGVCF and GATK GenotypeGVCFs. Typically preformed on a run/flowcell level then merged again accross all runs/flowcells
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:+UseParallelGC -XX:ParallelGCThreads=4 -Dsamjdk.buffer_size=4194304 -Xmx24G -jar $GATK_JAR \
--analysis_type CombineGVCFs -G StandardAnnotation -G StandardHCAnnotation \
--disable_auto_index_creation_and_locking_when_reading_rods \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--variant alignment/${SAMPLE1}/${SAMPLE1}.hc.g.vcf.gz \
--variant alignment/${SAMPLE2}/${SAMPLE2}.hc.g.vcf.gz \
--variant alignment/${SAMPLE_N}/${SAMPLE_N}.hc.g.vcf.gz \
--out variants/allSamples_run1.hc.g.vcf.gz
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=1048576 -Xmx30G -jar $GATK_JAR \
--analysis_type GenotypeGVCFs --useNewAFCalculator -G StandardAnnotation -G StandardHCAnnotation \
--disable_auto_index_creation_and_locking_when_reading_rods \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--variant variants/allSamples_run1.hc.g.vcf.gz \
--out variants/allSamples_run1.hc.vcf.gz
3.3 Jointed called variants from N samples are filtered using a variant recalibration step ``GATK Variant Quality Score Recalibration (VQSR)` which uses known, highly validated variant resources (omni, 1000 Genomes, hapmap). SNPs and INDELs are processed independently.
3.3.1 Generate tranches
SNP variant recalibration
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=1048576 -Xmx24G -jar $GATK_JAR \
--analysis_type VariantRecalibrator -nt 11 \
--disable_auto_index_creation_and_locking_when_reading_rods \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
-input variants/allSamples_run1.hc.vcf.gz \
-resource:hapmap,known=false,training=true,truth=true,prior=15.0 \ $MUGQIC_INSTALL_HOME_DEV/genomes/species/Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.hapmap_3.3.vcf.gz \
-resource:omni,known=false,training=true,truth=false,prior=12.0 \ $MUGQIC_INSTALL_HOME_DEV/genomes/species/Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.1000G_omni2.5.vcf.gz \
-resource:1000G,known=false,training=true,truth=false,prior=10.0 \ $MUGQIC_INSTALL_HOME_DEV/genomes/species/Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.1000G_phase1.snps.high_confidence.vcf.gz \
-resource:dbsnp,known=true,training=false,truth=false,prior=6.0 \ $MUGQIC_INSTALL_HOME_DEV/genomes/species/Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.dbSNP142.vcf.gz \
-an QD -an FS -an DP -an SOR -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 -mode SNP \
--recal_file variants/allSamples.hc.snps.recal \
--tranches_file variants/allSamples.hc.snps.tranches \
--rscript_file variants/allSamples.hc.snps.R
INDEL variant recalibration
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=1048576 -Xmx24G -jar $GATK_JAR \
--analysis_type VariantRecalibrator -nt 11 \
--disable_auto_index_creation_and_locking_when_reading_rods \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
-input variants/allSamples_run1.hc.vcf.gz \
-resource:mills,known=false,training=true,truth=true,prior=12.0 \ $MUGQIC_INSTALL_HOME/genomes/species/Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.Mills_and_1000G_gold_standard.indels.vcf.gz \
-resource:dbsnp,known=true,training=false,truth=false,prior=2.0 \ $MUGQIC_INSTALL_HOME_DEV/genomes/species/Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.dbSNP142.vcf.gz -an QD -an DP -an FS -an SOR \
-tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 -mode INDEL \
--recal_file variants/allSamples.hc.indels.recal \
--tranches_file variants/allSamples.hc.indels.tranches \
--rscript_file variants/allSamples.hc.indels.R
3.3.2 Apply tranches
SNPs
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=1048576 -Xmx24G -jar $GATK_JAR \
--analysis_type ApplyRecalibration -nt 11 \
--disable_auto_index_creation_and_locking_when_reading_rods \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
-input variants/allSamples_run1.hc.vcf.gz \
--ts_filter_level 99.5 -mode SNP \
--tranches_file variants/allSamples.hc.snps.tranches \
--recal_file variants/allSamples.hc.snps.recal \
--out variants/allSamples.hc.snps_raw_indels.vqsr.vcf.gz
INDELs
java -Djava.io.tmpdir=${SLURM_TMPDIR} -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=1048576 -Xmx24G -jar $GATK_JAR \
--analysis_type ApplyRecalibration -nt 11 \
--disable_auto_index_creation_and_locking_when_reading_rods \
--reference_sequence /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
-input variants/allSamples.hc.snps_raw_indels.vqsr.vcf.gz \
--ts_filter_level 99.0 -mode INDEL \
--tranches_file variants/allSamples.hc.indels.tranches \
--recal_file variants/allSamples.hc.indels.recal \
--out variants/allSamples.hc.vqsr.vcf.gz
Compute quality control metrics
A series of scripts compute several metrics derived from the output of the analysis above. Here are the full set of commands used to generate these metrics.
#alignment and insert size metrics
gatk --java-options "-XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx20G" \
CollectMultipleMetrics \
--PROGRAM=CollectAlignmentSummaryMetrics \
--PROGRAM=CollectInsertSizeMetrics \
--VALIDATION_STRINGENCY=SILENT \
--REFERENCE_SEQUENCE=/cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
--OUTPUT=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.all.metrics \
--MAX_RECORDS_IN_RAM=1000000
#OxoG metrics
gatk --java-options "-XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx20G" \
CollectOxoGMetrics \
--VALIDATION_STRINGENCY=SILENT \
--INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
--OUTPUT=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.oxog_metrics.txt \
--REFERENCE_SEQUENCE=/cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--MAX_RECORDS_IN_RAM=4000000
#GC biais metrics
gatk --java-options "-XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 -Xmx20G" \
CollectGcBiasMetrics \
--VALIDATION_STRINGENCY=SILENT \
--ALSO_IGNORE_DUPLICATES=TRUE \
--INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
--OUTPUT=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.qcbias_metrics.txt \
--CHART=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.qcbias_metrics.pdf \
--SUMMARY_OUTPUT=metrics/dna/${SAMPLE}/picard_metrics/${SAMPLE}.qcbias_summary_metrics.txt \
--REFERENCE_SEQUENCE=/cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa \
--MAX_RECORDS_IN_RAM=4000000
#qualimap metrics
qualimap bamqc -nt 2 \
-bam alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam -outdir metrics/dna/${SAMPLE}/qualimap \
--java-mem-size=55G
#bedGraph generation on mark duplication
bedtools genomecov -bga \
-ibam alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam > alignment/${SAMPLE}/${SAMPLE}.sorted.dup.BedGraph
#generate tdf file
java -Xmx8G -Djava.awt.headless=true -jar $IGVTOOLS_JAR count -f min,max,mean -w 25 \
alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
alignment/${SAMPLE}/${SAMPLE}.sorted.dup.tdf \
/cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/genome/Homo_sapiens.GRCh38.fa.fai
#verifyBAMid to estimate contamination
verifyBamID \
--vcf /cvmfs/soft.mugqic/CentOS6/genomes/species/Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.1000G_phase1.snps.high_confidence.EUR.vcf \
--bam alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
--out metrics/dna/${SAMPLE}/verifyBamId/${SAMPLE} \
--verbose --ignoreRG --noPhoneHome
#vcftools
vcftools --missing-indv \
--gzvcf variants/allSamples.hc.vcf.gz \
--out variants/allSamples
#vcftools
vcftools --depth \
--gzvcf variants/allSamples.hc.vcf.gz \
--out variants/allSamples
#GATK4 CrosscheckFingerprints for sample mixup detection
gatk --java-options "-Djava.io.tmpdir=${SLURM_TMPDIR} -XX:+UseParallelGC -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 \
-Dsamjdk.use_async_io_read_samtools=true -Dsamjdk.use_async_io_write_samtools=true -Dsamjdk.use_async_io_write_tribble=false -Xmx16G" \
CrosscheckFingerprints --NUM_THREADS 2 --EXIT_CODE_WHEN_MISMATCH 0 \
--VALIDATION_STRINGENCY SILENT \
--TMP_DIR ${SLURM_TMPDIR} \
--INPUT=alignment/${SAMPLE}/${SAMPLE}.sorted.dup.bam \
--HAPLOTYPE_MAP $MUGQIC_INSTALL_HOME/genomes/species/Homo_sapiens.GRCh38/annotations/Homo_sapiens.GRCh38.fingerprint.map \
--LOD_THRESHOLD 3.0 \
--OUTPUT=metrics/dna/sample.fingerprint
gatk --java-options "-Djava.io.tmpdir=${SLURM_TMPDIR} -XX:+UseParallelGC -XX:ParallelGCThreads=1 -Dsamjdk.buffer_size=4194304 \
-Dsamjdk.use_async_io_read_samtools=true -Dsamjdk.use_async_io_write_samtools=true -Dsamjdk.use_async_io_write_tribble=false -Xmx16G" \
ClusterCrosscheckMetrics \
--VALIDATION_STRINGENCY SILENT \
--TMP_DIR ${SLURM_TMPDIR} \
--INPUT metrics/dna/sample.fingerprint \
--LOD_THRESHOLD 5.0 \
--OUTPUT=metrics/dna/cluster.fingerprints
Reference Genome and Software Versions¶
Reference Genome: Genome Reference Consortium Human Build 38 patch release 13 (GenBank GCA_000001405.28)
Software versions
skewer 0.2.2
bwa 0.7.17
GATK 4.1.8.1 and 3.8
sambamba 0.7.0
igvtools 2.3.67
java 1.8.0_72
picard 2.23.3
python 2.7.14
R 3.5.1_3.7
samtools 1.9
bcftools 1.9
htslib 1.9
snpeff 4.3
qualimap 2.2.2dev
fastqc 0.11.5
verifyBAMid 1.1.3
bedtools 2.29.2
bvatools 1.6
vcftools 0.1.14